Explaining the emergence of complex self-organization and structures in living cells in terms of general physical mechanisms is an outstanding challenge. Astounding progress in experimental techniques integrated with ever-increasing computing power and sophisticated theoretical models are bringing us every day a step closer to tackling this challenge. It is becoming more and more apparent that the goal of quantitative biology is not anymore unreachable.
Artificial Intelligence driven scientific discovery of molecular mechanisms
Computational sciences are going through a fascinating time, offering possibilities that were not long ago unthinkable. At the same time, we are living a frustrating situation. On the one hand, ever-growing computational power and sophisticated software enable us to simulate increasingly complex systems over ever longer times. On the other hand, intrinsic limitations in our methods and upper bounds to the scalability of distributed calculations severely limit the sampling of exciting phenomena in extensive “brute force” simulations. Despite incredible technological advancements, simulating many complex molecular mechanisms is still out of reach. This first limitation is what we might call the “sampling problem.”
Our limitations in sampling are not the only problem. Already computational methodologies produce vast amounts of data to analyze, e.g., in the form of trajectories of biological systems simulated by molecular dynamics. These are generally describing the evolution of systems in very-high dimensions. Extracting quantitative human-friendly insight from these data is a critical challenge, which is mostly still tackled by labor-intensive and bias-prone visual inspection. Moreover, translating what has been learned from complex simulations into mathematical models is even more challenging. We call this second limitation the “learning problem.”
To summarize, simulating many interesting, complex systems is still impossible with straightforward approaches. Yet, at the same time, we struggle to learn from those systems that we can simulate already. Exascale supercomputers are going to further exacerbate the interpretation problem and, due to the slowing down of Moore’s law and the “Communication Wall,” fail to fully solve the sampling problem.
The interpretation and sampling problems are two faces of the same coin. Learning the essential features that describe a complex mechanism – its reaction coordinate — enables advanced sampling schemes that focus computational power on the simulation of the mechanism itself. The two problems should, therefore, be tackled at the same time. However, finding a suitable reaction coordinate is hugely challenging, even for relatively simple dynamical systems. Moreover, its search still almost entirely relies on human visual analysis. This dependence on human experts threatens to become one of the main bottlenecks in the exascale computing era. We, therefore, need to develop tools able to perform the same tasks with minimal human intervention.
Recently, I co-developed a groundbreaking artificial intelligence-driven computational framework, which integrates advanced sampling schemes, statistical inference, neural networks, and deep learning. This novel framework autonomously plans, performs, and learns quantitative mechanistic insight from computer simulations of complex molecular events. In a first proof-of-concept, this approach provided mathematical expressions describing how many- body collective solvent configurations control the dissociation of ions in water, a well-known, long-standing challenge in the field of theoretical chemistry. Our artificial intelligence-assisted computational framework is not only an enabling technology but also solves the problem of distributing a vast number of simulations on massively-parallel supercomputers. This achievement opens the door to exascale simulations of biological systems.
By using a combination of deep learning and symbolic regression, our AI autonomously distilled simple mathematical expressions from simulated data. These expressions quantitatively reproduced the reaction coordinate of complex molecular events. This result showcases the exciting possibility of building quantitative models in an automatic and data-driven way. Although these models are, in general, correlative, we are now working to build physical models automatically. Our long term goal is building AIs that derive deep physical insight from noisy data of stochastic systems.
Computational structural virology: SARS-CoV-2
The current Covid19 global pandemic painfully reminds us how vulnerable our lives and societies still are to harmful viruses. The spread of SARS-CoV-2 has changed the way we live and will continue to do so, to the extent unimaginable a few months ago. The development of an effective vaccine is probably the only way to neutralize this historic threat. Unfortunately, developing vaccines is still a formidable challenge. After more than 40 years of intense work, we do not have a vaccine against HIV yet!
In the case of SARS-CoV-2, an impressive number of vaccine developments are ongoing at breakneck speed. Early evidence from some trials encourages a more optimistic outlook, and we may have some first working vaccines within the next months. However, we still do not know how broad a protection these vaccines will provide or how easily the virus will develop resistance by mutating. A further worrisome possibility is that the emergence of new harmful vaccines will become an increasingly more frequent event. Ideally, we want to make the rational design of a vaccine easier and more efficient, and possibly create vaccines that offer protection against a broad family of viruses.
Viruses like SARS-CoV-2 and HIV protect their genetic material behind a lipid envelope and use particular proteins on their surfaces to target and infect human cells. These proteins are the harpoon used by the virus to attack cells, but at the same time, are they weak spots because they are among the few parts exposed to the immune system. In the case of SARS-CoV-2, this is the infamous spike protein. A popular strategy to develop a vaccine is to stimulate the immune system to produce neutralizing antibodies. These are molecules that bind to the virus in strategic points to hinder essential conformational changes or prevent binding to the host cell.
Modeling and simulations can facilitate the development of vaccines by revealing the dynamics of viral parts exposed to the immune system. Proteins like the spike of SARS-CoV-2 are highly mobile. Also, a layer of complex molecules called glycans cover the spike. Glycans are highly mobile, continually sweeping the surface of the spike, and preventing the binding of possible antibodies. Therefore, we think that glycans form a protective shield. Computer simulations put in motion atomistic structures resolved in experiments, and allow us to assess how dynamic and accessible the spike of SARS-CoV-2 is.
We recently developed an atomistic model of the spike of SARS-CoV-2 embedded in a realistic membrane. Although produced independently, the agreement with electron tomography imaging of in situ spikes is stunning. Our model aided the interpretation of these images, clarifying the structure of the glycan shields and explaining the dynamic behavior of the long stalk region that connects the spike to the viral envelope. Additionally, by carefully studying the surface of the spike, we identified possible regions that could be targeted by antibodies.
We are currently following a similar strategy to understand better other proteins of SARS-CoV-2 and the analog of the spike protein in HIV.
Molecular mechanisms of sensing and regulation in cellular membranes and the unfolded protein response
In cellular membranes, thousands of different lipids and membrane proteins self-assemble into spatially heterogeneous and temporally dynamic complexes. Membrane proteins and their host membranes are engaged in a constant dialogue: membranes tell proteins how to move and arrange, and proteins reshape membranes in return. The dynamic modulation of this intricate choreography is at the origin of many important regulatory phenomena in the cell.
One of the main questions of the lab is how cells sense and control the physical state of their membranes. In collaboration with experimental laboratories, we revealed the molecular mechanisms of four key membrane sensor proteins controlling fundamental cellular processes in the endoplasmic reticulum and the plasma membrane. In particular, MD simulations discovered a rotational conformational change in dimers of the transmembrane proteins MGA2 that, in yeast, senses an excess of saturated lipids in the ER.
In all eukaryotic cells, the formation of mesoscopic clusters of the membrane protein IRE1 activates the unfolded protein response (UPR). The UPR is a fundamental control program that protects the cell from a broad series of detrimental stresses. It plays a key role in development, cancer, neurodegeneration, chronic pain, and metabolic diseases. In the presence of stresses, IRE1 assembles into complexes and large clusters in the membrane of the endoplasmic reticulum. How this happens is still mysterious. Gaining a deep mechanistic understanding of the activation of the UPR is of fundamental biological and medical importance.
In a groundbreaking interdisciplinary collaboration, we showed how the conformation of the sensor protein IRE1 senses aberrant lipid compositions in the ER, paving the way to a mechanistic understanding of the membrane-based activation of the UPR.
Despite remarkable progress, we still lack a deep mechanistic understanding of how the molecular environment of the ER membrane controls the formation of assemblies and clusters of IRE1. AI-assisted simulations will be instrumental in establishing how different lipid compositions determine stability and life-time of complexes of IRE1. The challenge will be to integrate this information in a multi-scale reaction-diffusion model of the dynamics and morphology of clusters of IRE1 at the level of the whole ER and link the outcome to microscopy data. This work will not only serve as a template to understand other cellular control mechanism but also enable design of manipulative experiments.
Self-assembly and conformational changes of biomolecules
Protein folding is a paradigm of self-assembly that is still challenging to study by conventional simulations. My PhD revolved around the development of an advanced computer simulation framework based on a path integral formulation of diffusive dynamics, which vastly reduces the computational cost of studying protein folding in MD simulations. The sampling power of this framework enabled me to characterize the folding landscape of a WW-domain, which revealed that folding could proceed via several distinct mechanisms. The same approach permitted the first-ever reported folding simulation in atomistic resolution of a knotted protein, i.e., a protein that displays a topological knot in its native configuration, revealing how transient interactions can control the folding mechanism.
Exploring the immense configuration space of biomolecules is one of the outstanding challenges in simulations. In an interdisciplinary collaboration with applied mathematicians, I developed an advanced data-driven exploration method where machine learning guides and accelerates the exploration in MD simulations. Diffusion Maps – a powerful dimensionality reduction technique – extracts a low-dimensional representation of the region explored by simulated trajectories. New MD runs are started beyond this region, providing data for the following iteration. This work resulted in one of the first applications of machine learning to simulations of conformational changes and self-assembly of protein complexes in lipid membranes. It discovered a dissociation mechanism not seen in millisecond-long equilibrium simulations.
We are currently working to harness the power of machine learning further to simulate complex conformational changes. In particular, we focus on developing methods to simulate the spontaneous and reversible formation of assemblies of membrane proteins.
Statistical modeling of biophysical experiments and simulations
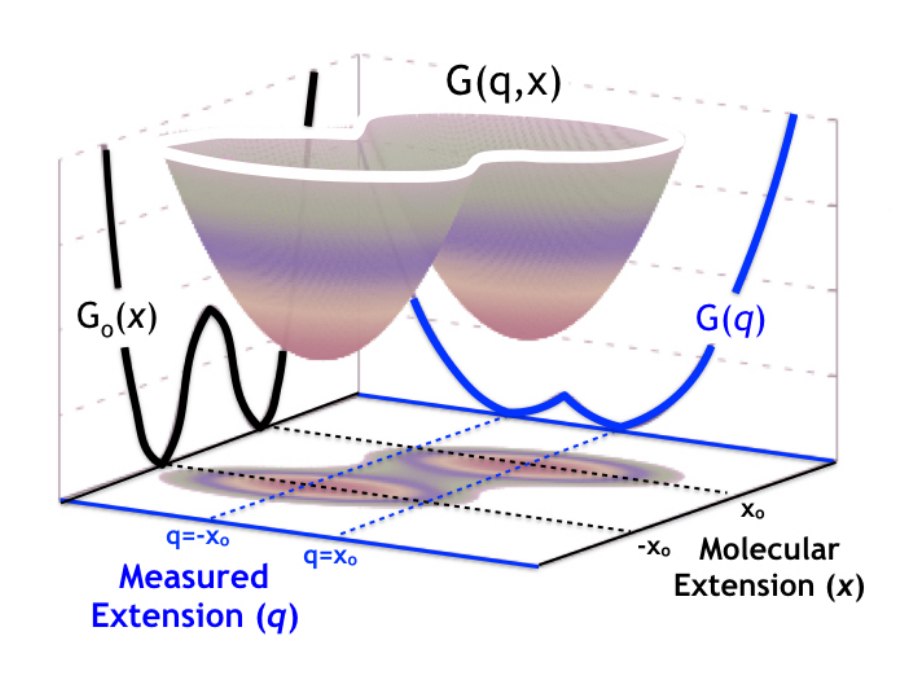
Single-molecule experiments and simulations give us unprecedented access to conformational changes in molecular systems. Interpreting the complex data produced by such techniques, however, is non-trivial. We, therefore, face the challenge of modeling these simulations and experiments to extract as much accurate information as possible. In particular, we must strive to understand and control the influence of the experimental apparatus on the molecular measurements. For instance, in protein or nucleic acid single-molecule force spectroscopy, the measuring device affects our knowledge of the dynamics of the molecular extension, which is, in this case, the relevant observable. In a recent collaboration, I developed a theoretical framework based on the statistical mechanics of diffusive models with hidden coordinates. This framework allows us to control the information that we can extract by measuring the splitting probability (committor) in constant force-spectroscopy experiments.
One area of research of the lab involves modeling data produced by simulations and experiments via Markov models with hidden degrees of freedom.